OTC Monograph Process FDA Reform

After two years of stalled progress on approving legislation for FDA reform of Over the Counter (OTC) Monograph Drug Review, Congress finally did so on March 27, 2020 under the largest stimulus package in the United States’ history, the Coronavirus Aid, Relief, and Economic Security Act (CARES Act).
The reform has been championed by the FDA and industry as a much-needed solution to advance and modernize the FDA’s approach to governing the regulatory process and to secure additional resources to oversee the growing category of nonprescription medicines. The new provisions are intended to provide drug manufacturers a less cumbersome and more streamlined, flexible framework to advance innovation while giving the FDA quicker avenues to develop and issue regulations and respond to drug safety concerns.
There are two sections to the new OTC Monograph Reform legislation, the first addressing the new FDA review process and the second addressing the new user fees that will be issued to drug sponsors and drug manufacturers. Before advancing to understand the details of these new provisions, a brief background on how drugs get classified as Over the Counter (OTC) is required.
FDA Reform Background
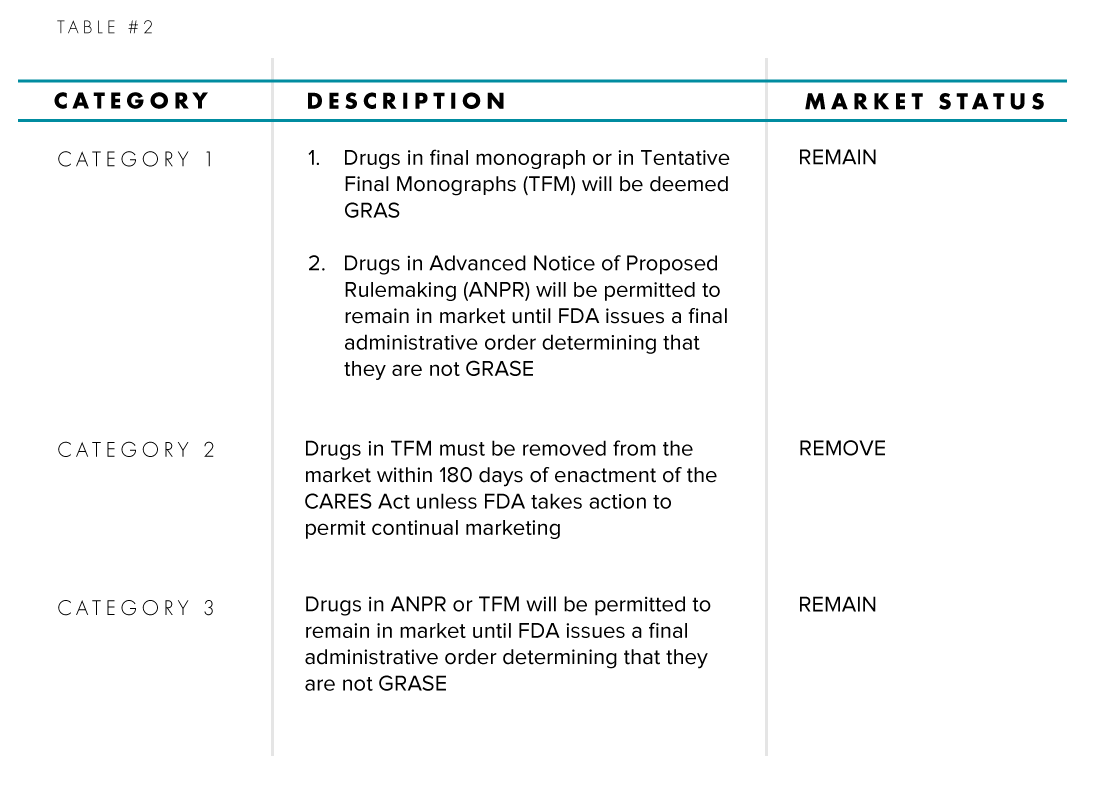
In 1972, the FDA initiated the OTC Drug Review, which was a comprehensive review of the safety, effectiveness, and labeling of all OTC medicines on the market. The Review brings together an array of industry, healthcare, and regulatory representatives to rigorously review the three categories for Active Pharmaceutical Ingredients (API) and determine via regulations which Active Pharmaceutical Ingredients are GRASE (Generally Recognized as Safe and Effective) for their intended use. Those that are classified as GRASE do not require an application to be reviewed by the FDA before they are commercialized while ones that aren’t GRASE require one. The three categories are summarized in Table #1:

The rule-making process is a three-phased approach in which each phase requires a Federal Registrar publication by the FDA. The end result establishes standards (monograph) for an OTC therapeutic class. The first phase brings together an advisory panel that evaluates the safety and effectiveness of Active Pharmaceutical Ingredients (API) in OTC drugs along with its claims. These conclusions are then published by the FDA in the form of an Advanced Notice of Proposed Rulemaking (ANPR) for public commentary. The second phase is the agency’s review of the public’s comments and new data that may have come available after the initial review. After the conclusion of this review, the FDA publishes a Tentative Final Monograph (TFM) which is again open for public commentary. The third and last phase is the final regulation in the form of a drug monograph. All drugs and indications that do not fall under a monograph require a premarket approval before they are commercialized.
Since 1972, there are marketed OTC drugs that have not been formalized into a final drug monograph. These medicines are still in an incomplete stage either under an Advanced Notice of Proposed Rulemaking (ANPR) or Tentative Final Monograph (TFM). The new Act will drive the backlog of these medicines to a final status.
The FDA Reform Review Process
The new legislation will establish Section 505G to the Federal Food, Drug, and Cosmetics Act (FDCA). Under this section, there are 17 subparts (a-q) that cover the new review process for OTC drugs ranging from the confidentiality of information submitted to the FDA to key definitions and terms used throughout 505G. Most industry attention will be focused on subparts (a)-(c).
Subpart (a) presents the new regulatory status of existing nonprescription medicines marketed under the OTC monograph process; subpart (b) describes changes to the three-phase rule making process; and subpart (c) provides the requirements for minor changes made to OTC drugs.
Focusing on Subpart (a), Table #2 enumerates the changes to a drug’s status based on its Active Pharmaceutical Ingredient (API) category and its place in the rule-making process phase.
To streamline and simplify the administrative order process, subpart (b) discusses three new procedures for the FDA and industry to issue orders. This will replace the cumbersome three-phase rule making process.
- FDA Initiated Order: this will entail public notice, opportunity for comment, dispute resolution procedures, and opportunity for administrative hearing, and for judicial review
- Expedited Orders: when drugs are deemed to present an immediate hazard to public health or when the FDA requires safety statements in labeling. These can be issued in final form before an opportunity to comment, dispute resolution, and a hearing
- Industry Initiated Orders: industry proceedings from sponsor submissions seeking approval of a new Active Pharmaceutical Ingredient (API), new dosage forms of existing APIs, or other changes to marketed products. The same procedures for FDA Initiated Orders will apply to Industry Initiated Orders.
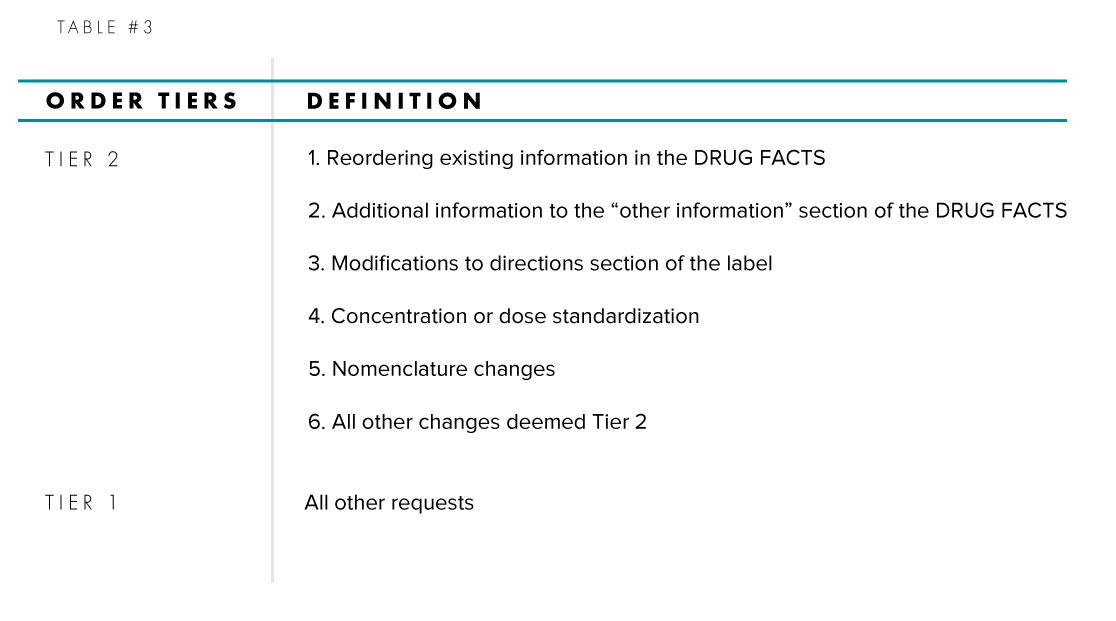
For Industry Initiated Orders, Table #3 summarizes the two tiers of order requests that can be submitted by a drug sponsor:
Minor changes in dosage forms to existing marketed OTC drugs that don’t require approval from the FDA before commercialization is codified in subpart (c); however, these change can only be executed by a drug sponsor once the FDA, via an FDA Initiated Order, establishes the criteria and data requirements for such changes of a particular dosage form. Sponsors would likely have to carry out additional efficacy and safety data studies on the new dosage form and notify the FDA of its intention to make a change but will not be required to submit an application to the FDA prior to marketing.
Industry User Fees
A user fee program has never been established for OTC monograph drugs like it has for New Drug Applications (NDA), Abbreviated New Drug Application (ANDA), Biologics License Application (BLA), and new or existing Medical Devices (e.g. Pre-Market Approvals, 510k, etc.). User fees from these applications have not been used to fund the regulatory resources required to govern the OTC monograph system. A user fee program for OTC drugs will be dedicated to its governance operation and will facilitate the modernization of the system, including timely review processes and expedited responses to safety issues.
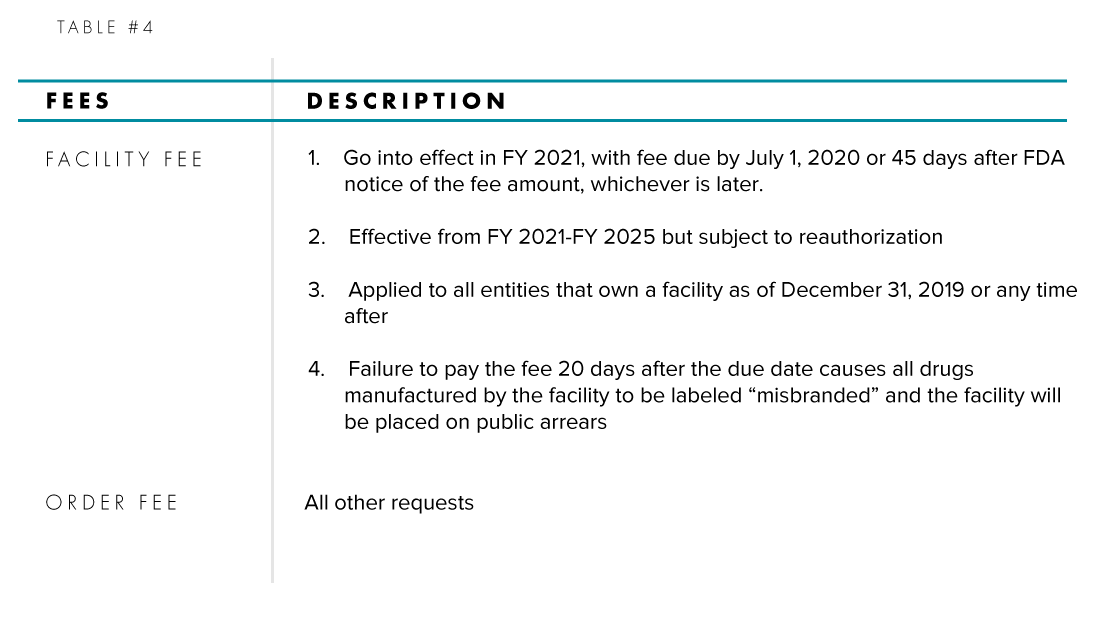
Two types of fees, Order Fees and Facility Fees, have been established with the new legislation and their details are summarized in Table #4.
Facility Fees will be applied annually to parties that own and operate an OTC drug manufacturing facility, whether the drug sponsor or a contract manufacturing organization (CMO). An OTC drug manufacturing facility is defined as a foreign or domestic entity at one geographic location or address engaged in the manufacturing or processing of the finished dosage form of an OTC monograph dru. A CMO is defined as a monograph drug facility where neither the owner nor any affiliate sells the OTC monograph drug produced at the facility directly to wholesalers, retailers, or consumers in the United States.
Other Major FDA Reform Provisions:
Three other major provisions of the Act deal with exclusivity, OTC sunscreen monographs, and Pediatric Indications for Cough and Cold Products. For exclusivity, a sponsor can gain this status over the marketing of the OTC drug if the industry order submitted is for a new Active Pharmaceutical Ingredient (API) or a change in an existing monograph based on new human data studies. The provision defines these studies as clinical trials of safety and effectiveness including actual use studies, pharmacokinetic studies, and bioavailability studies. Exclusivity would start on the date the requester may lawfully market a drug that is contingent on FDA’s final order
Separate OTC Sunscreen monographs provisions are codified in detail under the new legislation and dictates that all marketed sunscreens that adhere to the 1999 final monograph will be considered GRAS. It is to be recognized that the 1999 final monograph was never made effective in over 20 years; therefore, the new Act requires that the FDA revise the monograph within 18 months after enactment of the regulation and for it to become effective at least 1 year after publication.
As for pediatric indications for cough and cold dosing, the legislation will require the FDA to annually submit a letter to Congress describing its progress on evaluating monograph cough and cold products for children under the age of six until a final order is rendered. Until its administered, the annual progress letters from the FDA will continue.
Over the Counter (OTC) medicines have and will continue to be an integral part of the United States’ healthcare ecosystem. By 2025, the United States’ OTC market will be in excess of $44B or ~25% of the global market due to people’s growing tendency toward self-medication for minor diseases and the cost saving advantages for both patients and healthcare providers. The new legislation is a major step forward for both government and industry to lay a new foundation for a well-funded and streamlined process that assists the market’s projected growth, encourages industry innovation, mandates critical topics be addressed routinely, and provides faster methods for FDA to issue regulations and guidance.


